Milestone White Paper I
Jun 3, 2024

Abstract
Cryopreservation is a widely used technique for the storage of isolated neural cells and has recently been demonstrated to maintain electrical activity in simple neural organoids. However, recovery of action potentials from cryopreserved acutely resected neural tissue remains an ongoing challenge for the field. Here, we cryopreserve and rewarm acutely resected rat cerebellar slices, demonstrating electrical activity after rewarming. This is, to our knowledge, the first report of recovery of electrical activity in acutely resected mammalian brain tissue with accompanying protocols for validation and replication.
Main
Cryopreservation of isolated cells is a ubiquitous technique that is required for the storage and shipment of primary neural progenitor cells (Hancock et al, 2000). While neurons derived from these cells can provide some insights into single-cell biophysics and electrophysiology, their electrical activity is fundamentally different from neurons in the context of native microcircuits. For this reason, much neuroscience research is focused either in living organisms or in acute slices of tissue. Acute slices conveniently allow for electrical access to deep brain regions while maintaining much of the connectivity between cells (Frey et al, 2009). Given the difficulty in quickly accessing and maintaining the viability of human brain tissue for research use, the vast majority of these experiments are performed on murine tissue, limiting translatability (Day et al, 2013). An effective method for cryopreservation of acutely resected neural tissue from human donors would enable cryostorage and shipment of these valuable research samples, unlocking a new platform for basic neuroscience research and drug development. Recently, a protocol for successful cryopreservation of lab-grown brain organoids was shown, with impressive maintenance of structure and electrical activity after rewarming (Xue et al, 2024). However, the study did not demonstrate electrical activity in their cryopreserved sample of acutely resected neural tissue. Organoids, while invaluable for modeling aspects of disease and early brain development, face limitations compared to acute tissue slices, such as a lack of mature cellular organization and functional connectivity, which can impact their translational relevance in neuroscience research (Qian & Song, 2019).
Here, we present a method for cryopreservation of acute slices of rat cerebellar tissue, showing recovery of some electrical activity after cryopreservation and rewarming. This is, to our knowledge, the first demonstration of action potentials in cryopreserved and rewarmed acutely resected neural tissue.
Results
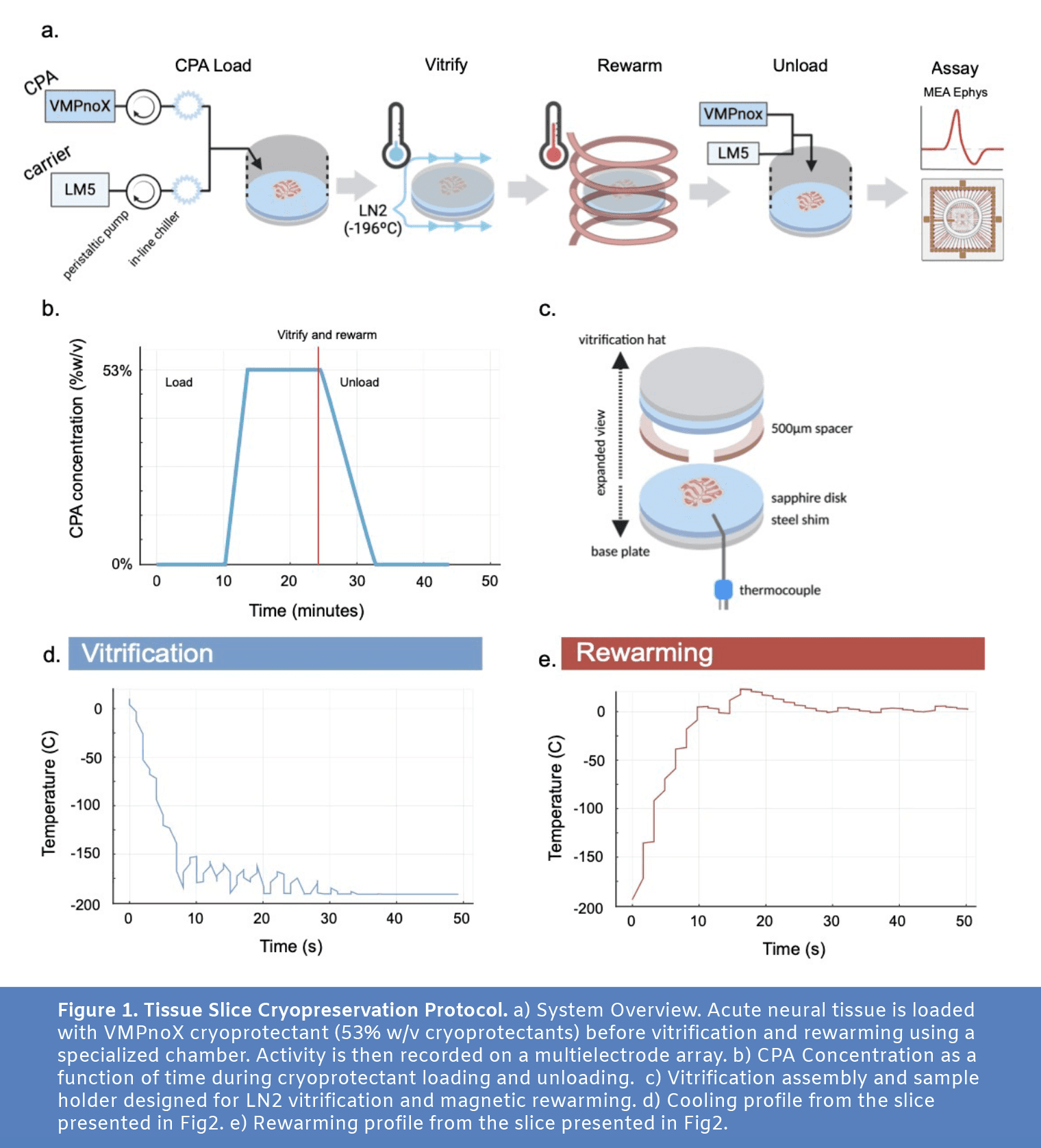
To achieve viable preservation, we first diffusively loaded 300 micron thick acute rat cerebellar slices with a cryoprotectant solution (Fig1a and Fig1b) whose composition is identical to the established VMP cryoprotectant (Han et al. 2023, Fahy et al. 2004) save the removal of one component (X-1000 ice blocker). In preparation for cryopreservation, acute slices of rat cerebellum were placed in a multifunctional sample holder with a custom well for diffusive loading of cryoprotectant (Fig4). The tissue was loaded with CPA, was rapidly cooled from 4ºC to -196ºC using a jet of liquid nitrogen (LN2), and then was rewarmed using an alternating magnetic field from an induction heater (Fig1d and Fig1e). The sample holder’s perfusion well was then placed back on top of the sample and cryoprotectant was unloaded using a linear ramp in perfused concentration into the well from the maximum CPA concentration to no CPA in 500 seconds. After ~45 minutes of post-cryopreservation incubation, the slices were transferred to a multielectrode array (3Brain Duplex) where activity was recorded at baseline before the addition of carbachol and the later addition of tetrodotoxin (Fig2).
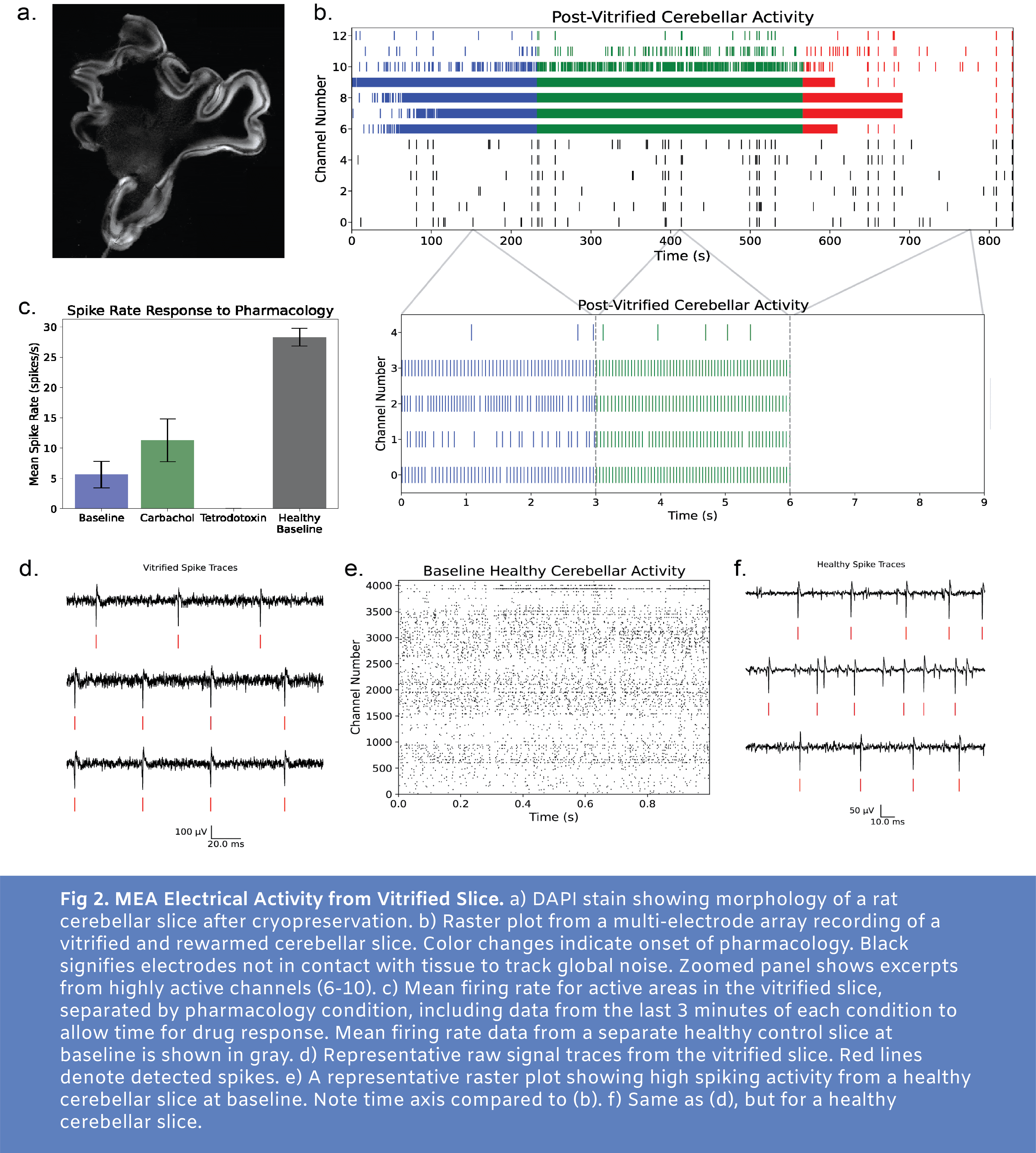
For N=4 slices tested with this protocol, activity was present and the signal responded as expected to pharmacology with carbachol increasing activity from baseline and tetrodotoxin silencing action potentials. In Figure 2, we present data from one of these slices (additional data available upon request, email hunter@cradle.xyz). At baseline, a number of electrodes detected high frequency spiking (Fig2b and Fig2c). With application of carbachol to increase neural excitability, an increase in mean spike rate was observed. With tetrodotoxin application, spiking was blocked, confirming that the earlier signal represented physiological spiking.
While neural activity was present in multiple MEA channels, it is clear that there is a marked reduction in electrical activity following this cryopreservation protocol. Sample data from a healthy control slice at baseline (no pharmacological stimulation) is shown in Fig2e.
Of the 4 cryopreserved slices with maintained electrical activity, the slice depicted in Fig2 showed no signs of macro cracking. Large thermal gradients applied across a volume of tissue after a phase change are likely to cause cracking. Future experiments will involve tuning the vitrification and rewarming profile aiming to completely avoid cracking while also cooling and warming fast enough to avoid damaging ice formation.
Discussion
Here, we demonstrate recovery of electrical activity in previously cryopreserved acute rodent neural tissue. Due to the high viscosity of vitrified water, bringing a sample to LN2 temperature (-196ºC) allows for virtually indefinite storage due to the extreme slowing of molecular motion, stopping metabolism and preventing ice formation. The finding that neural tissue can reach LN2 temperature and be rewarmed to recover electrical activity marks an important milestone on the trajectory to an acute slice preservation protocol appropriate for the storage of acutely resected human tissue for research applications.
Iterations and improvements to this protocol will be necessary to achieve a level of preservation and functional viability required for translatable research using this method. Continued screening of highly effective and minimally toxic CPA formulations specifically targeted for neural tissue will enable improved viability. Our next aim along this trajectory is to apply novel neural CPAs and thermal control protocols to demonstrate synaptic function and long-term potentiation (LTP) in an acute slice following cryopreservation, moving towards effective cryopreservation of slices that more closely resemble healthy controls.
Additionally, this work sheds light on the possibility of whole body reversible cryopreservation, which would enable medical hibernation to halt the progression of terminal diseases showing promise of a cure in the foreseeable future. We predict that neural tissue will pose a particular challenge compared to other organs due to limited capacity for regeneration, low redundancy, high osmotic sensitivity, and the presence of the blood brain barrier which may limit uptake of cryoprotective agents. Methods to translate technology tailored to the preservation of thin neural tissue sections to the preservation of an entire brain or organism will require a number of notable modifications. The CPA loading protocol must be compatible with perfusive loading through the vasculature, and the relevant CPAs must cross the blood brain barrier. In addition, sophisticated thermal approaches for homogenous rewarming techniques are needed.
It is not obvious which combination of approaches will yield an effective protocol that meets these and other complex needs, but as presented here, an acute neural slice platform demonstrating some electrical activity following cryopreservation presents a valuable jumping off point for future development and iteration.
Methods
All experiments were conducted in accordance with animal welfare and veterinary standards. The IACUC committee at Cradle Healthcare approved all studies.
Animals
CD (Sprague Dawley) rats were obtained from Charles River and housed with food and water ad libitum. Male and female rats aged postnatal day P12 to P19 were used for experiments.
Acute Slice Preparation
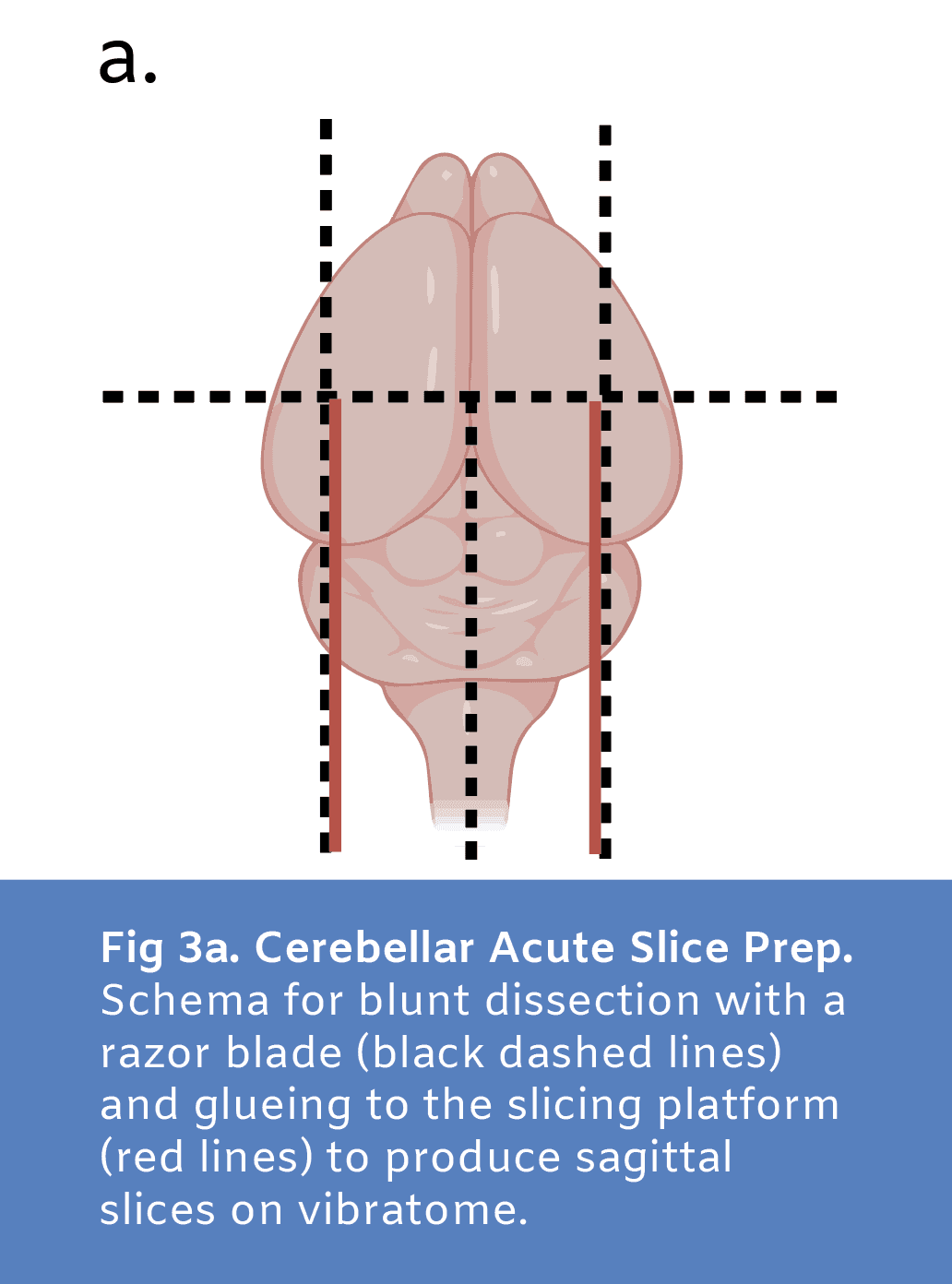
Acute sagittal cerebellar slices were prepared in order to evaluate viability of neural tissue following cryopreservation. Cerebellar slices were chosen due to high baseline activity compared to sections from other brain regions to allow for most sensitivity in our viability assays. A modified aCSF cutting solution (87mM sodium chloride, 25mM sodium bicarbonate, 1.25mM sodium phosphate monobasic, .5mM calcium chloride, 2.5mM potassium chloride, 25mM D-glucose, 75mM sucrose, 7mM magnesium chloride; Wang et al. 2024) containing high sucrose and magnesium to reduce activity and damage during slicing was prepared, osmolyzed and adjusted to 300 - 330 mOsm, and carbogenated with gas containing 95% oxygen and 5% carbon for at least 30 minutes prior to use. This solution was used for transcardiac perfusion, dissection, and vibratome slicing and was kept ice cold and carbogenated throughout. Following isoflurane anesthesia and transcardiac perfusion, the brain was dissected from the skull and glued onto the slicing platform against a block of 3% agarose for vibratome sectioning of sagittal cerebellar slices (Fig3a). 300μm thick sections were sliced and incubated in carbogenated aCSF solution (125mM sodium chloride, 25mM sodium bicarbonate, 1.25mM sodium phosphate monobasic, 2mM calcium chloride, 3mM potassium chloride, 25mM D-glucose, 1mM magnesium chloride, 3mM sodium pyruvate) at 32ºC for 30 minutes then transferred to carbogenated aCSF solution at room temperature for at least 15 minutes prior to experimentation.
CPA Solutions
The cryoprotectant solution VMP (16.8 wt% ethylene glycol, 22.3% DMSO, 12.9% formamide, 1% X-1000, 1% Z-1000) has been used previously in organ cryopreservation experiments (Han et al. 2023, Fahy et al. 2004). X-1000 and Z-1000 are synthetic ice blocker solutions obtained from 21st Century Medicine composed of 20% w/w% polyvinyl alcohol and 40% w/w% polyglycerol, respectively. In our experiments, we used a modification of VMP, here called VMPnoX (16.8 wt% ethylene glycol, 22.3% DMSO, 12.9% formamide, 1% Z-1000), made by simply removing X-1000 from the recipe for VMP. This solution was made by diluting in LM5 carrier solution (1mM adenine hydrochloride, 1mM calcium chloride, 90mM glucose, 5mM glutathione, 2mM magnesium chloride, 28.5mM potassium chloride, 7.2mM potassium phosphate monobasic, 10mM sodium bicarbonate, 45mM lactose, 45mM mannitol; Fahy et al. 2004). Solutions were made within 24 hours before use. For gradual loading and unloading of cryoprotectant in slices, the concentration of CPA was decreased by diluting with additional LM5 with programmatically controlled channels on an Ismatec Reglo ICC 4-channel peristaltic pump feeding into the perfusion well (Fig4b).
Hardware Design and Assembly Instructions
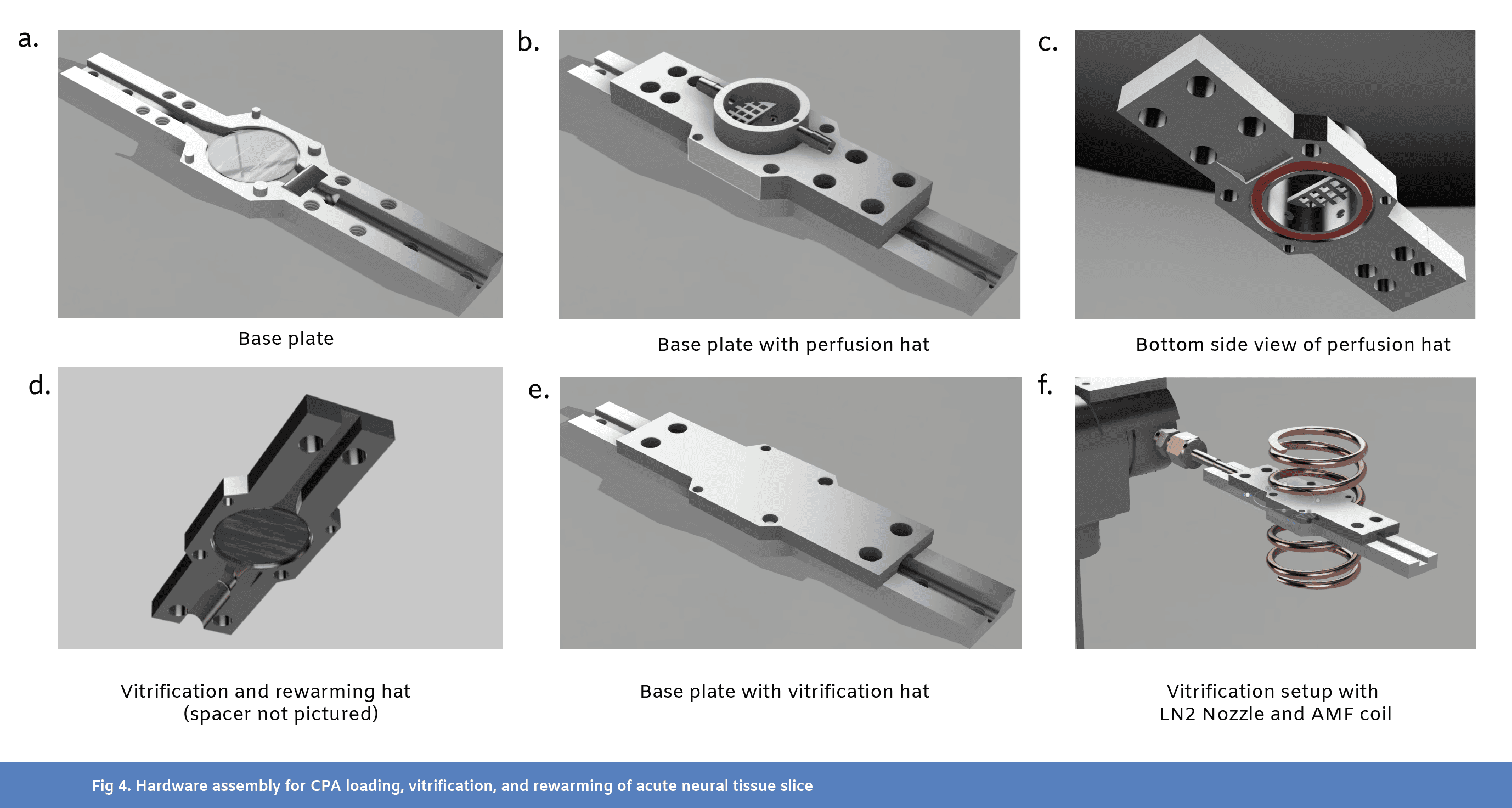
We designed 3 custom parts for this experiment (see CAD files), including a common base plate with two “hats” meant to support 1) loading and unloading of CPAs into a slice of tissue (Fig4b and Fig4c) and 2) vitrification and rewarming of the tissue (Fig4d-f). These parts were printed on a FormLabs 3B printer in Tough 1500 resin. For final assembly of the base plate (see CAD), a 20mm diameter, 500 µm thick double-sided-polish sapphire disk was attached to a 19mm diameter, 250 µm thick carbon-steel disk shim using MasterBond EP29LPSPAO-1 Black (Fig1c and Fig4a). This sapphire and steel wafer was epoxied into the base plate, leaving the sapphire side exposed.
The perfusion hat (see CAD) featured an inlet hose barb connection, leading into a hollow inner channel with four holes near the bottom of the well for fluid to enter (Fig4b and Fig4c). An outlet hose barb connection allowed fluid to be suctioned out of the well from the top. A mesh prevented tissue slices from being sucked into the outlet. An o-ring groove located at the bottom of the well allowed for a water tight seal against the sapphire wafer on the base plate, using an 18mm ID, 2mm thick o-ring.
The vitrification and rewarming hat (see CAD) was functionally a mirror of the base plate, with the same sapphire and steel wafer (Fig4d). However, a 500 µm, ½’’ ID PTFE spacer was epoxied to the sapphire shim on the vitrification hat to ensure that the tissue did not get squished in between the two wafers of sapphire (Fig1c). A small slot in the spacer was cut out to fit a thin, T-type thermocouple in the sandwich near the slice. The thermocouple was inserted from the rear (LN2 exhaust side). The tissue could then be sandwiched between the two wafers, and only come in contact with the sapphire.
For vitrification, a three-way solenoid valve was used to gate LN2 flow (Fig4f). When turned off, the valve was set to exhaust LN2 into an empty dewar. When turned on, the valve directed LN2 into the slice chamber. For rewarming, we used a 15 kW, 30-80 kHz induction heater and a coil with an ID of approximately 30mm for heating. A gap in the center of the coil was created to fit the slice chamber (Fig4f).
CPA Loading
Prior to CPA loading, the slice loading system described above was prepared, and the tubes were primed with LM5 carrier and VMPnoX CPA. Fresh cerebellar slices, prepared as described, were transferred to the perfusion well containing LM5 solution, and the CPA loading protocol was initiated. The loading protocol was as follows: 10 minutes of 0% w/v CPA (LM5 carrier only), 200 second ramp from 0% to 53% w/v CPA, hold at 53% w/v CPA for 10 minutes (Fig1b). The total flow rate into the inlet of the well was set to 2 mL/min and the flow rate at the outlet was set to 2.2 mL/min in order to maintain a steady fluid level within the well. The temperature setpoint throughout loading and unloading was 4ºC to 10ºC and was regulated by two inline glass heat exchangers. To improve temperature control, cryoprotectant loading was run in a refrigerator set to 4ºC with solution reservoirs on ice. To test toxicity of the cryoprotectant without contribution of damage associated with cryogenic cooling, a set of slices were run through the load unload protocol all the way through without pausing for vitrification and rewarming (data available upon request, email hunter@cradle.xyz).
Vitrification
At the end of the CPA loading protocol, slices were prepared for rapid cooling to cryogenic temperatures. Fluid was removed from the perfusion well and the slice was gently centered on the sapphire disk at the bottom of the well. The perfusion hat was removed and replaced with the vitrification hat (Fig4d, see CAD). The assembly was transferred to the vitrification and rewarming station, where it was hooked up to a pre-cooled LN2 nozzle for vitrification and nestled within the coil of an alternating magnetic field (AMF) induction heater for subsequent rewarming (Fig4f). LN2 was supplied from a 15-psi liquid nitrogen dewar. Once in place, a solenoid valve gating LN2 flow across the slice was opened to allow for rapid cooling. Temperature recording from the T-type thermocouple placed next to the slice was monitored, and LN2 flow was maintained for 30 seconds after minimum temperature (about -196ºC) had been reached. Cooling rates of 1140 ºC/min (calculated as an average over the period t=0 to t=10s, in Fig1d) were reached for the slice reported in Fig1 and Fig2, but this differed slightly from slice to slice.
AMF Rewarming
After holding at -196ºC for 1 minute (long enough to completely cool the sample to the endpoint temperature), flow of LN2 was halted and the sample was quickly rewarmed using an AMF electromagnet operating at 15W field strength and ~70kHz frequency. Warming occurred via the magnetic heating of two thin carbon steel disk shims attached to both sapphire disks on the side not in contact with the brain slice (Fig1c). A rate of 1280 ºC/min (calculated as an average over the period t=0 to t=10 seconds in Fig1e) was reached for the slice reported in Fig1 and Fig2. The temperature probe next to the slice monitored the temperature, and when the slice reached 0ºC, the AMF maintained the temperature at 0ºC with PID control.
CPA Unloading
After rewarming, the vitrification hat was removed from the assembly and the perfusion hat was re-installed. The CPA unloading protocol was initiated (Fig1b), which included a 500 second linear ramp from 53% to 0% w/v CPA, followed by 10 minutes of 0% w/v CPA (LM5 carrier only). At the end of the protocol, the slices were removed from the CPA perfusion device and placed in warm incubation in carbogenated aCSF for 30 minutes at 32ºC, followed by at least 15 minutes in carbogenated aCSF at room temperature.
MEA Slice Electrophysiology
Viability in tissue slices was evaluated by multi-electrode array electrophysiology using the 3Brain BioCAM DupleX system (Switzerland). Healthy control slices were evaluated soon after post-slicing incubation. Experimental slices were evaluated soon after post-unload incubation.
The 3Brain HD-MEA Acura 2D chip model was used, with 4,096 electrodes in a working area of 3.84mm x 3.84mm and an electrode size of 21 μm x 21 μm. Recordings were acquired with 20kHz sampling frequency. Slices were carefully placed on the multielectrode array chip and imaged under a stereoscope, secured with a silicon slice holder insert, and inserted into the BioCAM DupleX system for recording. The slice was perfused with carbogenated aCSF solution at ~2 mL/min, and the perfusion inlet and the chip base were held at 32ºC.
A contonuous recording was started and ~5 minutes of baseline activity was collected (Fig2b). Then, 50 uM carbachol was perfused over the slice for ~5 minutes. Carbachol is an acetylcholine receptor agonist and increases spiking and activity in cerebellar slices by activating muscarinic receptors, enhancing neuronal excitability and synaptic transmission (Pickford, 2019). Finally, 10 uM tetrodotoxin was perfused over the slice for ~5 minutes. Tetrodotoxin blocks neural activity by binding to and inhibiting voltage-gated sodium channels on neuronal membranes, preventing the initiation and propagation of action potentials (Wakita et al., 2015).
Data Analysis
Spike detection was performed in Brainwave 5 software (3Brain) using the Precise Timing Spike Detection (PTSD) algorithm, designed to identify extracellular neuronal spikes (Maccione et al., 2009). In PTSD, a local minimum is classified as a spike if it is followed within a given peak lifetime period by a local maximum (the overshoot), the peak-to-peak distance between the maximum and minimum is above a given multiple of the signal standard deviation (σ), and it occurs after a given refractory period following the previous spike. Here, the peak-to-peak threshold was set to 10σ with a peak lifetime period of 2 ms and a refractory period of 1 ms.
Histology
After MEA electrophysiology, we roughly visualized the morphology of one of the vitrified slices (Fig2a) with DAPI staining. After MEA electrophysiology, the slice was fixed for 20 minutes in 10% neutral buffered formalin and permeabilized in .3% Triton X-100 in PBS for 18 hours at 4ºC. The slice was then washed and stained with DAPI solution at 10μg/mL for 30 minutes to stain the nuclei of all cells. The slice was mounted on a slide in PBS and dehydrated in ethanol (10min at 30% EtOH, 10min 50%, 15 min 70%, 15min 80%, 20min 95%, 40min 100%), cleared in xylenes (40min), and mounted with a Vectamount Express Mounting Medium (Vector Labs). The slice was imaged on an Olympus FV3000 confocal microscope. A Z-stack of the whole slice was acquired with a 10x magnification objective (UPLXAPO10X), 1.5x zoom, and 405nm wavelength laser excitation.
References
Day, B., Stringer, B., Wilson, J., Jeffree, R., Jamieson, P., Ensbey, K., Bruce, Z., Inglis, P., Allan, S., Winter, C., Tollesson, G., Campbell, S., Lucas, P., Findlay, W., Kadrian, D., Johnson, D., Robertson, T., Johns, T., Bartlett, P., Osborne, G., & Boyd, A. (2013). Glioma Surgical Aspirate: A Viable Source of Tumor Tissue for Experimental Research. Cancers, 5, 357 - 371. https://doi.org/10.3390/cancers5020357.
Fahy GM, Wowk B, Wu J, Phan J, Rasch C, Chang A, Zendejas E. Cryopreservation of organs by vitrification: perspectives and recent advances. Cryobiology. 2004 Apr;48(2):157-78. doi: 10.1016/j.cryobiol.2004.02.002. Erratum in: Cryobiology. 2005 Jun;50(3):344. PMID: 15094092.
Frey, U., Egert, U., Heer, F., Hafizovic, S., & Hierlemann, A. (2009). Microelectronic system for high-resolution mapping of extracellular electric fields applied to brain slices.. Biosensors & bioelectronics, 24 7, 2191-8 . https://doi.org/10.1016/j.bios.2008.11.028.
Han, Z., Rao, J.S., Gangwar, L. et al. Vitrification and nanowarming enable long-term organ cryopreservation and life-sustaining kidney transplantation in a rat model. Nat Commun 14, 3407 (2023). https://doi.org/10.1038/s41467-023-38824-8
Hancock, C., Wetherington, J., Lambert, N., & Condie, B. (2000). Neuronal differentiation of cryopreserved neural progenitor cells derived from mouse embryonic stem cells.. Biochemical and biophysical research communications, 271 2, 418-21 . https://doi.org/10.1006/BBRC.2000.2631.
Maccione, A., Gandolfo, M., Massobrio, P., Novellino, A., Martinoia, S., & Chiappalone, M. (2009). A novel algorithm for precise identification of spikes in extracellularly recorded neuronal signals. Journal of Neuroscience Methods, 177(1), 241–249. https://doi.org/10.1016/j.jneumeth.2008.09.026
Pickford J, Apps R, Bashir ZI. Muscarinic Receptor Modulation of the Cerebellar Interpositus Nucleus In Vitro. Neurochem Res. 2019; 44(3): 627–635. doi: 10.1007/s11064-018-2613-9
Wakita, M., Kotani, N., & Akaike, N. (2015). Tetrodotoxin abruptly blocks excitatory neurotransmission in mammalian CNS.. Toxicon : official journal of the International Society on Toxinology, 103, 12-8 . https://doi.org/10.1016/j.toxicon.2015.05.003.
Wang C, Derderian KD, Hamada E, Zhou X, Nelson AD, Kyoung H, Ahituv N, Bouvier G, Bender KJ. Impaired cerebellar plasticity hypersensitizes sensory reflexes in SCN2A-associated ASD. Neuron. 2024 May 1;112(9):1444-1455.e5. doi: 10.1016/j.neuron.2024.01.029. Epub 2024 Feb 26. PMID: 38412857; PMCID: PMC11065582.
Xue W, Li H, Xu J, Yu X, Liu L, Liu H, Zhao R, Shao Z. (2024) Effective cryopreservation of human brain tissue and neural organoids. Cell Reports Methods. May; 4(5). https://doi.org/10.1016/j.crmeth.2024.100777
Qian X, Song H, Ming GL. Brain organoids: advances, applications and challenges. Development. 2019 Apr 16;146(8):dev166074. doi: 10.1242/dev.166074. PMID: 30992274; PMCID: PMC6503989.
Abstract
Cryopreservation is a widely used technique for the storage of isolated neural cells and has recently been demonstrated to maintain electrical activity in simple neural organoids. However, recovery of action potentials from cryopreserved acutely resected neural tissue remains an ongoing challenge for the field. Here, we cryopreserve and rewarm acutely resected rat cerebellar slices, demonstrating electrical activity after rewarming. This is, to our knowledge, the first report of recovery of electrical activity in acutely resected mammalian brain tissue with accompanying protocols for validation and replication.
Main
Cryopreservation of isolated cells is a ubiquitous technique that is required for the storage and shipment of primary neural progenitor cells (Hancock et al, 2000). While neurons derived from these cells can provide some insights into single-cell biophysics and electrophysiology, their electrical activity is fundamentally different from neurons in the context of native microcircuits. For this reason, much neuroscience research is focused either in living organisms or in acute slices of tissue. Acute slices conveniently allow for electrical access to deep brain regions while maintaining much of the connectivity between cells (Frey et al, 2009). Given the difficulty in quickly accessing and maintaining the viability of human brain tissue for research use, the vast majority of these experiments are performed on murine tissue, limiting translatability (Day et al, 2013). An effective method for cryopreservation of acutely resected neural tissue from human donors would enable cryostorage and shipment of these valuable research samples, unlocking a new platform for basic neuroscience research and drug development. Recently, a protocol for successful cryopreservation of lab-grown brain organoids was shown, with impressive maintenance of structure and electrical activity after rewarming (Xue et al, 2024). However, the study did not demonstrate electrical activity in their cryopreserved sample of acutely resected neural tissue. Organoids, while invaluable for modeling aspects of disease and early brain development, face limitations compared to acute tissue slices, such as a lack of mature cellular organization and functional connectivity, which can impact their translational relevance in neuroscience research (Qian & Song, 2019).
Here, we present a method for cryopreservation of acute slices of rat cerebellar tissue, showing recovery of some electrical activity after cryopreservation and rewarming. This is, to our knowledge, the first demonstration of action potentials in cryopreserved and rewarmed acutely resected neural tissue.
Results
To achieve viable preservation, we first diffusively loaded 300 micron thick acute rat cerebellar slices with a cryoprotectant solution (Fig1a and Fig1b) whose composition is identical to the established VMP cryoprotectant (Han et al. 2023, Fahy et al. 2004) save the removal of one component (X-1000 ice blocker). In preparation for cryopreservation, acute slices of rat cerebellum were placed in a multifunctional sample holder with a custom well for diffusive loading of cryoprotectant (Fig4). The tissue was loaded with CPA, was rapidly cooled from 4ºC to -196ºC using a jet of liquid nitrogen (LN2), and then was rewarmed using an alternating magnetic field from an induction heater (Fig1d and Fig1e). The sample holder’s perfusion well was then placed back on top of the sample and cryoprotectant was unloaded using a linear ramp in perfused concentration into the well from the maximum CPA concentration to no CPA in 500 seconds. After ~45 minutes of post-cryopreservation incubation, the slices were transferred to a multielectrode array (3Brain Duplex) where activity was recorded at baseline before the addition of carbachol and the later addition of tetrodotoxin (Fig2).
For N=4 slices tested with this protocol, activity was present and the signal responded as expected to pharmacology with carbachol increasing activity from baseline and tetrodotoxin silencing action potentials. In Figure 2, we present data from one of these slices (additional data available upon request, email hunter@cradle.xyz). At baseline, a number of electrodes detected high frequency spiking (Fig2b and Fig2c). With application of carbachol to increase neural excitability, an increase in mean spike rate was observed. With tetrodotoxin application, spiking was blocked, confirming that the earlier signal represented physiological spiking.
While neural activity was present in multiple MEA channels, it is clear that there is a marked reduction in electrical activity following this cryopreservation protocol. Sample data from a healthy control slice at baseline (no pharmacological stimulation) is shown in Fig2e.
Of the 4 cryopreserved slices with maintained electrical activity, the slice depicted in Fig2 showed no signs of macro cracking. Large thermal gradients applied across a volume of tissue after a phase change are likely to cause cracking. Future experiments will involve tuning the vitrification and rewarming profile aiming to completely avoid cracking while also cooling and warming fast enough to avoid damaging ice formation.
Discussion
Here, we demonstrate recovery of electrical activity in previously cryopreserved acute rodent neural tissue. Due to the high viscosity of vitrified water, bringing a sample to LN2 temperature (-196ºC) allows for virtually indefinite storage due to the extreme slowing of molecular motion, stopping metabolism and preventing ice formation. The finding that neural tissue can reach LN2 temperature and be rewarmed to recover electrical activity marks an important milestone on the trajectory to an acute slice preservation protocol appropriate for the storage of acutely resected human tissue for research applications.
Iterations and improvements to this protocol will be necessary to achieve a level of preservation and functional viability required for translatable research using this method. Continued screening of highly effective and minimally toxic CPA formulations specifically targeted for neural tissue will enable improved viability. Our next aim along this trajectory is to apply novel neural CPAs and thermal control protocols to demonstrate synaptic function and long-term potentiation (LTP) in an acute slice following cryopreservation, moving towards effective cryopreservation of slices that more closely resemble healthy controls.
Additionally, this work sheds light on the possibility of whole body reversible cryopreservation, which would enable medical hibernation to halt the progression of terminal diseases showing promise of a cure in the foreseeable future. We predict that neural tissue will pose a particular challenge compared to other organs due to limited capacity for regeneration, low redundancy, high osmotic sensitivity, and the presence of the blood brain barrier which may limit uptake of cryoprotective agents. Methods to translate technology tailored to the preservation of thin neural tissue sections to the preservation of an entire brain or organism will require a number of notable modifications. The CPA loading protocol must be compatible with perfusive loading through the vasculature, and the relevant CPAs must cross the blood brain barrier. In addition, sophisticated thermal approaches for homogenous rewarming techniques are needed.
It is not obvious which combination of approaches will yield an effective protocol that meets these and other complex needs, but as presented here, an acute neural slice platform demonstrating some electrical activity following cryopreservation presents a valuable jumping off point for future development and iteration.
Methods
All experiments were conducted in accordance with animal welfare and veterinary standards. The IACUC committee at Cradle Healthcare approved all studies.
Animals
CD (Sprague Dawley) rats were obtained from Charles River and housed with food and water ad libitum. Male and female rats aged postnatal day P12 to P19 were used for experiments.
Acute Slice Preparation
Acute sagittal cerebellar slices were prepared in order to evaluate viability of neural tissue following cryopreservation. Cerebellar slices were chosen due to high baseline activity compared to sections from other brain regions to allow for most sensitivity in our viability assays. A modified aCSF cutting solution (87mM sodium chloride, 25mM sodium bicarbonate, 1.25mM sodium phosphate monobasic, .5mM calcium chloride, 2.5mM potassium chloride, 25mM D-glucose, 75mM sucrose, 7mM magnesium chloride; Wang et al. 2024) containing high sucrose and magnesium to reduce activity and damage during slicing was prepared, osmolyzed and adjusted to 300 - 330 mOsm, and carbogenated with gas containing 95% oxygen and 5% carbon for at least 30 minutes prior to use. This solution was used for transcardiac perfusion, dissection, and vibratome slicing and was kept ice cold and carbogenated throughout. Following isoflurane anesthesia and transcardiac perfusion, the brain was dissected from the skull and glued onto the slicing platform against a block of 3% agarose for vibratome sectioning of sagittal cerebellar slices (Fig3a). 300μm thick sections were sliced and incubated in carbogenated aCSF solution (125mM sodium chloride, 25mM sodium bicarbonate, 1.25mM sodium phosphate monobasic, 2mM calcium chloride, 3mM potassium chloride, 25mM D-glucose, 1mM magnesium chloride, 3mM sodium pyruvate) at 32ºC for 30 minutes then transferred to carbogenated aCSF solution at room temperature for at least 15 minutes prior to experimentation.
CPA Solutions
The cryoprotectant solution VMP (16.8 wt% ethylene glycol, 22.3% DMSO, 12.9% formamide, 1% X-1000, 1% Z-1000) has been used previously in organ cryopreservation experiments (Han et al. 2023, Fahy et al. 2004). X-1000 and Z-1000 are synthetic ice blocker solutions obtained from 21st Century Medicine composed of 20% w/w% polyvinyl alcohol and 40% w/w% polyglycerol, respectively. In our experiments, we used a modification of VMP, here called VMPnoX (16.8 wt% ethylene glycol, 22.3% DMSO, 12.9% formamide, 1% Z-1000), made by simply removing X-1000 from the recipe for VMP. This solution was made by diluting in LM5 carrier solution (1mM adenine hydrochloride, 1mM calcium chloride, 90mM glucose, 5mM glutathione, 2mM magnesium chloride, 28.5mM potassium chloride, 7.2mM potassium phosphate monobasic, 10mM sodium bicarbonate, 45mM lactose, 45mM mannitol; Fahy et al. 2004). Solutions were made within 24 hours before use. For gradual loading and unloading of cryoprotectant in slices, the concentration of CPA was decreased by diluting with additional LM5 with programmatically controlled channels on an Ismatec Reglo ICC 4-channel peristaltic pump feeding into the perfusion well (Fig4b).
Hardware Design and Assembly Instructions
We designed 3 custom parts for this experiment (see CAD files), including a common base plate with two “hats” meant to support 1) loading and unloading of CPAs into a slice of tissue (Fig4b and Fig4c) and 2) vitrification and rewarming of the tissue (Fig4d-f). These parts were printed on a FormLabs 3B printer in Tough 1500 resin. For final assembly of the base plate (see CAD), a 20mm diameter, 500 µm thick double-sided-polish sapphire disk was attached to a 19mm diameter, 250 µm thick carbon-steel disk shim using MasterBond EP29LPSPAO-1 Black (Fig1c and Fig4a). This sapphire and steel wafer was epoxied into the base plate, leaving the sapphire side exposed.
The perfusion hat (see CAD) featured an inlet hose barb connection, leading into a hollow inner channel with four holes near the bottom of the well for fluid to enter (Fig4b and Fig4c). An outlet hose barb connection allowed fluid to be suctioned out of the well from the top. A mesh prevented tissue slices from being sucked into the outlet. An o-ring groove located at the bottom of the well allowed for a water tight seal against the sapphire wafer on the base plate, using an 18mm ID, 2mm thick o-ring.
The vitrification and rewarming hat (see CAD) was functionally a mirror of the base plate, with the same sapphire and steel wafer (Fig4d). However, a 500 µm, ½’’ ID PTFE spacer was epoxied to the sapphire shim on the vitrification hat to ensure that the tissue did not get squished in between the two wafers of sapphire (Fig1c). A small slot in the spacer was cut out to fit a thin, T-type thermocouple in the sandwich near the slice. The thermocouple was inserted from the rear (LN2 exhaust side). The tissue could then be sandwiched between the two wafers, and only come in contact with the sapphire.
For vitrification, a three-way solenoid valve was used to gate LN2 flow (Fig4f). When turned off, the valve was set to exhaust LN2 into an empty dewar. When turned on, the valve directed LN2 into the slice chamber. For rewarming, we used a 15 kW, 30-80 kHz induction heater and a coil with an ID of approximately 30mm for heating. A gap in the center of the coil was created to fit the slice chamber (Fig4f).
CPA Loading
Prior to CPA loading, the slice loading system described above was prepared, and the tubes were primed with LM5 carrier and VMPnoX CPA. Fresh cerebellar slices, prepared as described, were transferred to the perfusion well containing LM5 solution, and the CPA loading protocol was initiated. The loading protocol was as follows: 10 minutes of 0% w/v CPA (LM5 carrier only), 200 second ramp from 0% to 53% w/v CPA, hold at 53% w/v CPA for 10 minutes (Fig1b). The total flow rate into the inlet of the well was set to 2 mL/min and the flow rate at the outlet was set to 2.2 mL/min in order to maintain a steady fluid level within the well. The temperature setpoint throughout loading and unloading was 4ºC to 10ºC and was regulated by two inline glass heat exchangers. To improve temperature control, cryoprotectant loading was run in a refrigerator set to 4ºC with solution reservoirs on ice. To test toxicity of the cryoprotectant without contribution of damage associated with cryogenic cooling, a set of slices were run through the load unload protocol all the way through without pausing for vitrification and rewarming (data available upon request, email hunter@cradle.xyz).
Vitrification
At the end of the CPA loading protocol, slices were prepared for rapid cooling to cryogenic temperatures. Fluid was removed from the perfusion well and the slice was gently centered on the sapphire disk at the bottom of the well. The perfusion hat was removed and replaced with the vitrification hat (Fig4d, see CAD). The assembly was transferred to the vitrification and rewarming station, where it was hooked up to a pre-cooled LN2 nozzle for vitrification and nestled within the coil of an alternating magnetic field (AMF) induction heater for subsequent rewarming (Fig4f). LN2 was supplied from a 15-psi liquid nitrogen dewar. Once in place, a solenoid valve gating LN2 flow across the slice was opened to allow for rapid cooling. Temperature recording from the T-type thermocouple placed next to the slice was monitored, and LN2 flow was maintained for 30 seconds after minimum temperature (about -196ºC) had been reached. Cooling rates of 1140 ºC/min (calculated as an average over the period t=0 to t=10s, in Fig1d) were reached for the slice reported in Fig1 and Fig2, but this differed slightly from slice to slice.
AMF Rewarming
After holding at -196ºC for 1 minute (long enough to completely cool the sample to the endpoint temperature), flow of LN2 was halted and the sample was quickly rewarmed using an AMF electromagnet operating at 15W field strength and ~70kHz frequency. Warming occurred via the magnetic heating of two thin carbon steel disk shims attached to both sapphire disks on the side not in contact with the brain slice (Fig1c). A rate of 1280 ºC/min (calculated as an average over the period t=0 to t=10 seconds in Fig1e) was reached for the slice reported in Fig1 and Fig2. The temperature probe next to the slice monitored the temperature, and when the slice reached 0ºC, the AMF maintained the temperature at 0ºC with PID control.
CPA Unloading
After rewarming, the vitrification hat was removed from the assembly and the perfusion hat was re-installed. The CPA unloading protocol was initiated (Fig1b), which included a 500 second linear ramp from 53% to 0% w/v CPA, followed by 10 minutes of 0% w/v CPA (LM5 carrier only). At the end of the protocol, the slices were removed from the CPA perfusion device and placed in warm incubation in carbogenated aCSF for 30 minutes at 32ºC, followed by at least 15 minutes in carbogenated aCSF at room temperature.
MEA Slice Electrophysiology
Viability in tissue slices was evaluated by multi-electrode array electrophysiology using the 3Brain BioCAM DupleX system (Switzerland). Healthy control slices were evaluated soon after post-slicing incubation. Experimental slices were evaluated soon after post-unload incubation.
The 3Brain HD-MEA Acura 2D chip model was used, with 4,096 electrodes in a working area of 3.84mm x 3.84mm and an electrode size of 21 μm x 21 μm. Recordings were acquired with 20kHz sampling frequency. Slices were carefully placed on the multielectrode array chip and imaged under a stereoscope, secured with a silicon slice holder insert, and inserted into the BioCAM DupleX system for recording. The slice was perfused with carbogenated aCSF solution at ~2 mL/min, and the perfusion inlet and the chip base were held at 32ºC.
A contonuous recording was started and ~5 minutes of baseline activity was collected (Fig2b). Then, 50 uM carbachol was perfused over the slice for ~5 minutes. Carbachol is an acetylcholine receptor agonist and increases spiking and activity in cerebellar slices by activating muscarinic receptors, enhancing neuronal excitability and synaptic transmission (Pickford, 2019). Finally, 10 uM tetrodotoxin was perfused over the slice for ~5 minutes. Tetrodotoxin blocks neural activity by binding to and inhibiting voltage-gated sodium channels on neuronal membranes, preventing the initiation and propagation of action potentials (Wakita et al., 2015).
Data Analysis
Spike detection was performed in Brainwave 5 software (3Brain) using the Precise Timing Spike Detection (PTSD) algorithm, designed to identify extracellular neuronal spikes (Maccione et al., 2009). In PTSD, a local minimum is classified as a spike if it is followed within a given peak lifetime period by a local maximum (the overshoot), the peak-to-peak distance between the maximum and minimum is above a given multiple of the signal standard deviation (σ), and it occurs after a given refractory period following the previous spike. Here, the peak-to-peak threshold was set to 10σ with a peak lifetime period of 2 ms and a refractory period of 1 ms.
Histology
After MEA electrophysiology, we roughly visualized the morphology of one of the vitrified slices (Fig2a) with DAPI staining. After MEA electrophysiology, the slice was fixed for 20 minutes in 10% neutral buffered formalin and permeabilized in .3% Triton X-100 in PBS for 18 hours at 4ºC. The slice was then washed and stained with DAPI solution at 10μg/mL for 30 minutes to stain the nuclei of all cells. The slice was mounted on a slide in PBS and dehydrated in ethanol (10min at 30% EtOH, 10min 50%, 15 min 70%, 15min 80%, 20min 95%, 40min 100%), cleared in xylenes (40min), and mounted with a Vectamount Express Mounting Medium (Vector Labs). The slice was imaged on an Olympus FV3000 confocal microscope. A Z-stack of the whole slice was acquired with a 10x magnification objective (UPLXAPO10X), 1.5x zoom, and 405nm wavelength laser excitation.
References
Day, B., Stringer, B., Wilson, J., Jeffree, R., Jamieson, P., Ensbey, K., Bruce, Z., Inglis, P., Allan, S., Winter, C., Tollesson, G., Campbell, S., Lucas, P., Findlay, W., Kadrian, D., Johnson, D., Robertson, T., Johns, T., Bartlett, P., Osborne, G., & Boyd, A. (2013). Glioma Surgical Aspirate: A Viable Source of Tumor Tissue for Experimental Research. Cancers, 5, 357 - 371. https://doi.org/10.3390/cancers5020357.
Fahy GM, Wowk B, Wu J, Phan J, Rasch C, Chang A, Zendejas E. Cryopreservation of organs by vitrification: perspectives and recent advances. Cryobiology. 2004 Apr;48(2):157-78. doi: 10.1016/j.cryobiol.2004.02.002. Erratum in: Cryobiology. 2005 Jun;50(3):344. PMID: 15094092.
Frey, U., Egert, U., Heer, F., Hafizovic, S., & Hierlemann, A. (2009). Microelectronic system for high-resolution mapping of extracellular electric fields applied to brain slices.. Biosensors & bioelectronics, 24 7, 2191-8 . https://doi.org/10.1016/j.bios.2008.11.028.
Han, Z., Rao, J.S., Gangwar, L. et al. Vitrification and nanowarming enable long-term organ cryopreservation and life-sustaining kidney transplantation in a rat model. Nat Commun 14, 3407 (2023). https://doi.org/10.1038/s41467-023-38824-8
Hancock, C., Wetherington, J., Lambert, N., & Condie, B. (2000). Neuronal differentiation of cryopreserved neural progenitor cells derived from mouse embryonic stem cells.. Biochemical and biophysical research communications, 271 2, 418-21 . https://doi.org/10.1006/BBRC.2000.2631.
Maccione, A., Gandolfo, M., Massobrio, P., Novellino, A., Martinoia, S., & Chiappalone, M. (2009). A novel algorithm for precise identification of spikes in extracellularly recorded neuronal signals. Journal of Neuroscience Methods, 177(1), 241–249. https://doi.org/10.1016/j.jneumeth.2008.09.026
Pickford J, Apps R, Bashir ZI. Muscarinic Receptor Modulation of the Cerebellar Interpositus Nucleus In Vitro. Neurochem Res. 2019; 44(3): 627–635. doi: 10.1007/s11064-018-2613-9
Wakita, M., Kotani, N., & Akaike, N. (2015). Tetrodotoxin abruptly blocks excitatory neurotransmission in mammalian CNS.. Toxicon : official journal of the International Society on Toxinology, 103, 12-8 . https://doi.org/10.1016/j.toxicon.2015.05.003.
Wang C, Derderian KD, Hamada E, Zhou X, Nelson AD, Kyoung H, Ahituv N, Bouvier G, Bender KJ. Impaired cerebellar plasticity hypersensitizes sensory reflexes in SCN2A-associated ASD. Neuron. 2024 May 1;112(9):1444-1455.e5. doi: 10.1016/j.neuron.2024.01.029. Epub 2024 Feb 26. PMID: 38412857; PMCID: PMC11065582.
Xue W, Li H, Xu J, Yu X, Liu L, Liu H, Zhao R, Shao Z. (2024) Effective cryopreservation of human brain tissue and neural organoids. Cell Reports Methods. May; 4(5). https://doi.org/10.1016/j.crmeth.2024.100777
Qian X, Song H, Ming GL. Brain organoids: advances, applications and challenges. Development. 2019 Apr 16;146(8):dev166074. doi: 10.1242/dev.166074. PMID: 30992274; PMCID: PMC6503989.